
ÜTS
TİTCK Yeni ÜTS Kayıt Kılavuzu Ne Anlama Geliyor? Tıbbi Cihaz Firmaları İçin Operasyonel Etkiler
TCKKD-KLVZ-03 ile yürürlüğe giren yeni ÜTS Kayıt Kılavuzunun firma kaydı, belge kaydı, cihaz başvurusu, MDR ve IVDR geçiş hükümleri ile toplu işlemler üzerindeki operasyonel etkileri. Türkiye’deki tıbbi cihaz firmaları için kapsamlı yol haritası.
- Yazar
- UTSBridge Ekibi
- Yayın tarihi
- 18 Mayıs 2026
- Okuma süresi
- 13 dk okuma
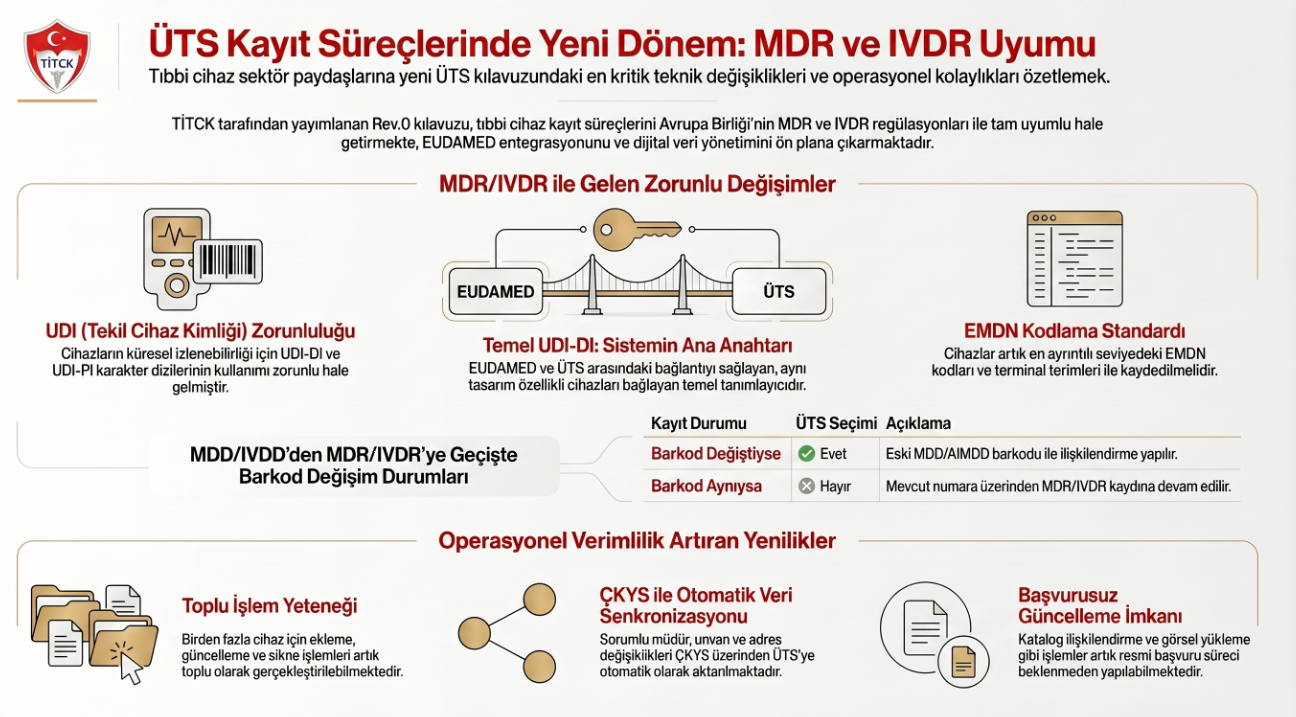
Bu yazı bir tanıtım metni değildir. TCKKD-KLVZ-03 kılavuzunun tıbbi cihaz firmaları için doğuracağı kayıt, belge ve başvuru etkilerini operasyonel açıdan ele alır. Resmi kılavuzun yerine geçmez; ekiplerin hazırlık yapmasına yardımcı olur.
Türkiye’de tıbbi cihaz sektörünün en kritik operasyonlarından biri Ürün Takip Sistemi kayıt süreçleridir. Ürün kaydı, belge bildirimi, firma yetkisi, ithalatçı bilgisi, uygunluk beyanı, sertifika, sistem ve işlem paketi kaydı, geçiş hükümleri ve toplu bildirimler birbirinden bağımsız gibi görünse de pratikte aynı zincirin parçalarıdır.
TİTCK tarafından yayımlanan "Tıbbi Cihazların Ürün Takip Sistemine Kayıt Süreçlerine İlişkin Kılavuz" bu zinciri yeniden daha net, daha kapsamlı ve daha operasyonel hale getiriyor. Bu kılavuz küçük bir açıklama metni değil. ÜTS’de ürün ve belge kaydı yapan firmaların günlük işleyişini doğrudan etkileyecek bir ana referans doküman niteliğinde.
Kılavuzun en önemli mesajı şu: ÜTS kaydı artık sadece ürün bilgisi girmekten ibaret görülmemeli. Ürün kaydı, belge doğruluğu, firma rolü, mevzuat rejimi, MDR ve IVDR uyumu, geçiş hükümleri, uygunluk değerlendirme prosedürü ve başvuru süreci birlikte yönetilmesi gereken bir operasyon haline geliyor.
Bu kılavuz neden önemli?
Yeni kılavuzun amacı, tıbbi cihaz yönetmelikleri doğrultusunda kayıt süreçlerinin nasıl uygulanacağını belirlemek. Kapsamı yalnızca ürün kaydıyla sınırlı değil. Tıbbi cihazlara ilişkin belge kayıt işlemleri, firma kayıt işlemleri ve ÜTS’de yapılacak diğer kayıt bildirim işlemleri de bu kapsama dahil.
Bu nedenle kılavuz, firmalar için sadece mevzuat ekibinin okuyacağı bir belge değil. Ürün yönetimi, kalite, regülasyon, ithalat, satış operasyonları, depo, stok, ERP ve ÜTS bildirim süreçlerini ilgilendiren ortak bir çalışma alanı oluşturuyor.
Asıl değişim burada: ÜTS’ye bilgi girmek bir ekran işi olmaktan çıkıyor, firmanın ürün verisini, belge arşivini, yetki bilgisini ve başvuru takibini düzenli tutmasını gerektiren bir süreç haline geliyor.
Eski kılavuz dönemi kapanıyor
Kılavuzun son hükümlerinde, 15/11/2022 tarihinde yayımlanan TCKKD-KLVZ-01 doküman numaralı "Tıbbi Cihazların Ürün Takip Sistemi Kaydına İlişkin Kılavuz Rev.1" adlı kılavuzun yürürlükten kaldırıldığı belirtiliyor.
Bu çok önemli bir ayrım. Çünkü burada sadece eski metnin üzerine birkaç satır eklenmiş değil. ÜTS kayıt süreçleri, yeni yönetmelikler, geçiş hükümleri, belge iş kuralları ve toplu işlem başlıklarıyla birlikte daha geniş bir çerçeveye taşınıyor.
Eski yönetmelik, yeni yönetmelik ayrımı artık daha kritik
Kılavuz, eski tıbbi cihaz yönetmelikleri ile yeni tıbbi cihaz yönetmeliklerini ayrı ele alıyor. Eski tarafta MDD, AIMDD ve IVDD var. Yeni tarafta MDR ve IVDR var. İçindekiler bölümünde eski yönetmelikler kapsamında cihaz ve SİP kayıtları ayrı, yeni yönetmelikler kapsamında MDR cihaz, MDR SİP, MDR ITC ve IVDR cihaz kayıtları ayrı başlıklar altında düzenlenmiş.
Bu ayrım operasyonel olarak çok şey değiştiriyor. Bir ürünün sadece GTIN’i, ürün adı veya markası yeterli değil. Ürünün hangi yönetmelik rejimine tabi olduğu, hangi sınıfa girdiği, hangi belgeye dayandığı ve geçiş hükümlerinden yararlanıp yararlanmadığı bilinmek zorunda.
Yanlış rejim seçimi, yanlış belge bağlantısı veya eksik sınıflandırma, ürünün ÜTS’de kayıt sürecinin uzamasına veya başvurunun olumsuz sonuçlanmasına neden olabilir.
Belge yönetimi artık ürün kaydının kalbi haline geliyor
Kılavuzda belge türleri çok açık şekilde ayrılıyor. EC Sertifikası eski yönetmelikler için, AB Sertifikası MDR veya IVDR için kullanılıyor. Aynı ayrım uygunluk beyanları için de geçerli. Eski yönetmelik kapsamında düzenlenen beyanlar ile MDR veya IVDR kapsamındaki AB Uygunluk Beyanları aynı mantıkla ele alınmıyor.
Bu, firmalar için büyük bir operasyonel uyarı demek. Ürünün belgesi sadece "PDF var mı?" diye kontrol edilemez. Belgenin türü, kapsamı, tarihi, imalatçı bilgisi, yetkili temsilci bilgisi, sertifika numarası, onaylanmış kuruluş bilgisi, ürün kapsamı ve ilgili yönetmelik ile uyumu birlikte kontrol edilmelidir.
Kılavuz ayrıca AB Uygunluk Beyanı için Temel UDI-DI, ürün adı, ürün kodu, katalog numarası, kullanım amacı ve izlenebilirliği sağlayan bilgileri de vurguluyor. Bu da ürün kartı ile belge arasında güçlü bir veri bağı kurulması gerektiğini gösteriyor.
Pratikte şu tablo ortaya çıkıyor: Belge arşivi dağınık olan, sertifika bitiş tarihlerini takip etmeyen, belge kapsamını ürün bazında eşleştirmeyen firmalar ÜTS kayıtlarında daha fazla sorun yaşayacak.
Kullanma kılavuzu ve Türkçe doküman konusu daha hassas hale geliyor
Kılavuzda belge diline ve Türkçe doküman yüklemelerine ilişkin detaylar da önemli. Orijinal belge yabancı dildeyse Türkçe doküman alanına yeminli tercüman tarafından yapılan Türkçe tercümenin eksiksiz yüklenmesi gerektiği belirtiliyor. Orijinal belge Türkçe veya Türkçe dahil çok dilli ise farklı bir yükleme mantığı uygulanıyor.
Bu nokta özellikle ithalatçı firmalar için önemli. Çünkü ürünün teknik belgesi, kullanma kılavuzu, sertifikası veya beyanı yabancı dilde olabilir. Bu durumda sadece dosyayı sisteme yüklemek yetmez. Belgenin doğru alana, doğru dil seçimiyle, doğru tercüme niteliğiyle yüklenmesi gerekir.
Operasyonel risk şudur: Firma ürün kaydını teknik olarak tamamladığını düşünür, fakat belge dil yapısı veya tercüme içeriği nedeniyle başvuru süreci uzayabilir.
MDR kayıtlarında UDI ve Temel UDI-DI öne çıkıyor
MDR kapsamındaki ürünlerde ürün kimliği artık çok daha güçlü bir veri disiplinine bağlı. UDI-DI, Temel UDI-DI, ürün tanımı, kullanım amacı, GMDN, EMDN, sınıf, uygunluk değerlendirme prosedürü ve belge ilişkileri birlikte düşünülmeli.
Bu, özellikle ürün portföyü geniş olan firmalar için ciddi bir veri kalitesi ihtiyacı doğuruyor. Bir ürünün ticari adı ile katalog numarası, GTIN’i, Temel UDI-DI’si, belge kapsamı ve kullanım amacı birbirinden kopuk tutulursa, ÜTS kaydı sırasında çelişkiler ortaya çıkabilir.
Kılavuzun eklerinde AB Uygunluk Beyanı örneklerinde katalog veya referans numarası, ürün adı veya tanımı, kullanım amacı, GMDN kodu, EMDN kodu, sınıf veya sınıflandırma kuralı ve Temel UDI-DI gibi alanların birlikte yer alması bu yaklaşımı güçlendiriyor.
Firmalar için net sonuç: MDR ürünleri için ürün ana verisi artık yalnızca satış veya stok kartı mantığıyla yönetilemez. Regülasyon verisi ürün kartının ayrılmaz parçası haline gelmelidir.
IVDR tarafında sınıf ve test türü ayrımları daha görünür hale geliyor
IVDR kapsamındaki cihazlarda Sınıf A, Sınıf A Steril, Sınıf B, Sınıf C, Sınıf D, kişisel test cihazı, hastabaşı test cihazı ve destek tanı cihazı gibi ayrımlar kritik hale geliyor.
Kılavuz, IVDR belge iş kurallarında bu ayrımları ayrıca ele alıyor. Örneğin Sınıf D cihazlar, kişisel test cihazları, hastabaşı test cihazları ve destek tanı cihazları için farklı eklerde yer alan belge iş kurallarının uygulanacağı belirtiliyor.
Bu durum özellikle in vitro tanı cihazı portföyü olan firmalar için önemli. Çünkü IVDR altında ürünün sadece "IVD ürün" olarak tanımlanması yeterli değil. Sınıfı, testin kullanım şekli, hasta başı mı kişisel test mi olduğu ve destek tanı niteliği belge gerekliliklerini etkileyebilir.
Operasyonel karşılığı açık: IVDR ürünleri için satış, ithalat ve kalite ekipleri aynı ürün sınıflandırmasını kullanmalıdır. Aksi halde belge eksikliği, yanlış beyan veya yanlış başvuru riski doğar.
SİP ve ITC kayıtları ayrı dikkat istiyor
Kılavuzda MDR kapsamında tıbbi cihaz kaydı yanında SİP ve ITC kayıtları da ayrı başlıklar altında yer alıyor. Bu, sistem veya işlem paketi oluşturan ya da ısmarlama tıbbi cihaz süreçleri olan firmaların bu alanları ayrı bir operasyon olarak ele alması gerektiğini gösteriyor.
SİP tarafında hata riski özellikle yüksektir. Çünkü burada sadece tek bir ürünün kaydı değil, birden fazla ürünün sistem veya işlem paketi mantığıyla birlikte yönetilmesi söz konusudur. Paketin içeriği, paketi oluşturan firma, SİP beyanı, belge kapsamı ve ürünlerin izlenebilirliği birlikte değerlendirilmelidir.
Bu nedenle SİP süreçleri manuel liste veya Excel üzerinden takip edildiğinde hata ihtimali artar. Bir ürünün pakete dahil edilmesi, paketten çıkarılması veya farklı bir ürünle değiştirilmesi, sadece ticari bir değişiklik değil, ÜTS kayıt ve mevzuat etkisi olan bir değişikliktir.
Toplu işlemler artık daha planlı yönetilmeli
Kılavuzun önemli başlıklarından biri de toplu işlemler. Eski yönetmelik, MDR ve IVDR kapsamında toplu cihaz ekleme veya güncelleme; toplu cihaz silme; başvurusuz cihaz güncelleme; piyasaya arz bilgisi ekleme; saklama veya kullanım koşulu ekleme ve ölçü bilgisi ekleme gibi işlemler ayrı ayrı ele alınmış.
Bu, firmalar için iki mesaj taşıyor. Birincisi, ÜTS toplu işlem mantığı artık operasyonun ayrılmaz parçası. İkincisi, toplu işlem yapmak veri kalitesi sorumluluğunu azaltmıyor, tam tersine artırıyor.
Toplu Excel yüklemelerinde bir alanın yanlış doldurulması yüzlerce ürünü etkileyebilir. Özellikle MDR ve IVDR ürünlerinde sınıf, UDI, EMDN, belge ilişkisi, saklama koşulu ve ölçü bilgisi gibi alanlar yanlışsa, hata tek ürünle sınırlı kalmaz.
Ürün kaydı başvuru ile tamamlanıyor
Kılavuzda belge başvurusu ve tıbbi cihaz başvurusu ayrı başlıklar altında ele alınıyor. Toplu ölçü bilgisi ekleme maddesinde bile ilgili kayıt veya güncelleme bildiriminin yönetmeliklere ve kılavuz hükümlerine uygunluğunun değerlendirilmesi için Kuruma cihaz başvurusu yapılması gerektiği belirtiliyor.
Bu nokta çok kritik. ÜTS’de veri girmek ile ürünün kayıtlı duruma gelmesi aynı şey değildir. Firma ürün bilgilerini hazırlayabilir, belgeyi yükleyebilir, Excel’i oluşturabilir; fakat başvuru süreci ve Kurum değerlendirmesi ayrıca takip edilmelidir.
Operasyonel etki şudur: Firmalar ürün kartı, belge durumu ve başvuru durumu arasında net ayrım yapmalıdır. "Verisi girildi", "başvuruya hazır", "başvuru yapıldı", "revizyon istendi", "kayıtlı" gibi statüler birbirine karıştırılmamalıdır.
İthal ürün veri değişikliği ayrı bir operasyon haline geliyor
Kılavuzda ithal ürün veri değişikliği talepleri ve bu taleplerin ÜTS’de oluşturulması ayrı bir bölüm olarak yer alıyor.
Bu başlık, ithalatçı firmalar için özellikle önemli. Çünkü ithal ürünlerde verinin kaynağı çoğu zaman yabancı imalatçı, yetkili temsilci, ithalatçı ve ÜTS’deki mevcut kayıtlar arasında dağılır. Ürün adı, marka, belge, katalog numarası, imalatçı bilgisi veya teknik veri değişikliklerinde kimin hangi veriyi güncelleyeceği ve bu güncellemenin nasıl başvuruya dönüşeceği net yönetilmelidir.
Buradaki risk şudur: Firma kendi iç sisteminde ürünü günceller ama ÜTS verisi aynı kalır. Ya da ÜTS’de bir düzeltme gerekir ama bunun başvuru süreci izlenmez. Bu da satış, stok, belge ve bildirim süreçlerinde çelişki yaratabilir.
MDR geçiş hükümleri firmaları doğrudan etkileyecek
Kılavuzda AB 2023/607 sayılı Tüzük hükümleri kapsamında MDR’ye geçiş hükümleri ve bu geçişten etkilenen cihaz veya belgeler için EBS ve ÜTS’de yapılacak başvurular ayrı bir bölüm olarak düzenlenmiş.
Bu bölümün firmalar açısından anlamı çok büyük. Çünkü bazı ürünler eski sertifika veya uygunluk beyanı ile geçiş sürecinden yararlanabilir. Ancak bu kendiliğinden olan bir durum değildir. Kılavuzda EBS başvurusu, ÜTS’de süre uzatımı talebi ve ilgili takip numarasının eklenmesi gibi süreçlerden bahsediliyor.
Yani geçiş süreci sadece "belgenin süresi uzadı" diye okunmamalıdır. Hangi belge için uzatım istendiği, hangi başvurunun yapıldığı, Kurumun neyi uygun gördüğü ve ÜTS’de bunun nasıl tanımlandığı takip edilmelidir.
IVDR geçiş hükümleri ve tedarik riski
IVDR tarafında AB 2024/1860 sayılı Tüzük özellikle yüksek riskli IVD cihazların piyasada bulunamama riskinin azaltılması, EUDAMED’in kademeli uygulanması ve tedarik kesintilerinin önceden bildirilmesi gibi başlıkları içeriyor. Kılavuz bu çerçeveyi IVDR geçiş hükümleri içinde ele alıyor.
Bu yalnızca regülasyon departmanının takip edeceği bir tarih meselesi değildir. Tedarik, ithalat, satış ve müşteri planlamasını doğrudan ilgilendirir.
Özellikle yüksek riskli IVD ürünlerinde geçiş tarihi, uygunluk değerlendirme durumu, onaylanmış kuruluş süreci, belge geçerliliği ve başvuru kanıtları birlikte takip edilmelidir. Bir ürünün kritik sağlık hizmetlerinde kullanılıyor olması, tedarik kesintisi riskini daha hassas hale getirir.
Firmalar bu yüzden IVDR geçişini sadece "son tarih ne?" sorusuyla değil, "hangi ürün, hangi sınıf, hangi belge, hangi başvuru, hangi tedarik riski?" sorularıyla yönetmelidir.
Sınıflandırma ve uygunluk değerlendirme prosedürleri daha fazla gündeme gelecek
Kılavuzun ilerleyen bölümlerinde cihazların sınıflandırılması, sınıflandırma kuralları ve uygunluk değerlendirme prosedürleri ayrıca ele alınmış. MDD, AIMDD, IVDD, MDR ve IVDR kapsamında sınıflandırma ve uygunluk değerlendirme prosedürleri ayrı başlıklar halinde yer alıyor.
Bu, sahada çok önemli bir değişim yaratır. Çünkü ürün sınıfı sadece teknik dosyada yazan bir bilgi değildir. ÜTS’de hangi belge iş kuralının çalışacağını, hangi belge türünün isteneceğini, hangi başvuru sürecinin işleyeceğini ve hangi geçiş hükmünün uygulanabileceğini etkiler.
Yanlış sınıflandırma, zincirleme hata üretir. Yanlış belge bağlanır, yanlış Excel hazırlanır, yanlış başvuru yapılır, yanlış statü takip edilir. Bu yüzden ürün sınıflandırması artık ürün master verisinin merkezinde yer almalıdır.
Belge iş kuralları firmalar için yeni kontrol listesi olmalı
Kılavuzda MDD, AIMDD, IVDD, MDR ve IVDR kapsamında ÜTS belge iş kuralları ayrı ayrı ele alınmış. IVDR tarafında Sınıf D, kişisel test, hastabaşı test ve destek tanı cihazları için farklı eklerin uygulanması gerektiği özellikle belirtiliyor.
Bu başlık, firmalar için altın değerinde. Çünkü hangi ürün için hangi belgenin gerektiği çoğu zaman kişisel tecrübeye veya eski alışkanlıklara göre yönetiliyor. Oysa yeni kılavuz, bu kararların daha sistematik verilmesini gerektiriyor.
Bir ürünün sınıfı değişirse belge gerekliliği değişebilir. Ürün MDR veya IVDR kapsamına geçerse belge türü değişebilir. Eski sertifika ile yeni AB sertifikası birbirine karıştırılırsa başvuru hatalı olabilir.
Bu nedenle firmaların ürün bazında bir belge uygunluk matrisi oluşturması gerekir. Her ürün için şu sorular net cevaplanmalıdır: Hangi yönetmelik? Hangi sınıf? Hangi belge türü? Hangi belge numarası? Hangi bitiş tarihi? Hangi ürünleri kapsıyor? ÜTS belge kaydı yapıldı mı? Başvuru sonucu ne?
Sahada ne değişecek?
Bu kılavuzun sahadaki etkisi birkaç alanda net hissedilecek.
- Ürün kayıt süreçleri daha fazla ön hazırlık isteyecek. Eksik belgeyle veya yarım ürün verisiyle ÜTS’ye gidip "sonra tamamlarız" yaklaşımı daha riskli hale gelecek.
- Kalite ve regülasyon ekiplerinin ürün master verisine daha fazla dahil olması gerekecek. Ürün adı, GTIN, katalog numarası, marka ve açıklama tek başına yeterli olmayacak. Temel UDI-DI, UDI-DI, EMDN, GMDN, sınıf, kullanım amacı, belge kapsamı ve geçiş bilgileri de aynı disiplinle tutulmalı.
- İthalatçı firmalarda imalatçıdan gelen belgelerin takibi daha kritik olacak. Yabancı dilde belge, tercüme, e-imzalı taahhütname, yetkili temsilci bilgisi ve belge kapsamı kontrol edilmeden başvuruya ilerlemek operasyonu yavaşlatabilir.
- Toplu işlemler daha fazla dikkat isteyecek. Çok sayıda ürün için Excel hazırlamak kolaylık sağlar ama aynı zamanda büyük ölçekli hata riski taşır.
- Geçiş hükümleri ürün bazında takip edilmeli. MDR veya IVDR geçişinden yararlanabilecek ürünlerin belge, EBS başvurusu, ÜTS talebi ve süre uzatımı kanıtları ayrı ayrı izlenmeli.
Firmalar şimdi ne yapmalı?
- Ürün portföyünü yönetmelik rejimine göre ayırın. Hangi ürün eski yönetmelik kapsamında, hangisi MDR kapsamında, hangisi IVDR kapsamında, hangisi SİP veya ITC sürecine giriyor netleştirilmeli.
- Belge envanterini temizleyin. EC Sertifikası, AB Sertifikası, Uygunluk Beyanı, AB Uygunluk Beyanı, SİP Beyanı, kullanma kılavuzu, ISO 13485, yetkili distribütörlük belgesi ve katalog gibi dokümanlar ürünlerle eşleştirilmeli.
- Kritik ürün verilerini kontrol edin. GTIN, katalog numarası, ürün adı, marka, kullanım amacı, GMDN, EMDN, UDI-DI, Temel UDI-DI, sınıf ve belge kapsamı arasında tutarsızlık olmamalı.
- Geçiş hükümlerinden yararlanan ürünleri ayrı listeye alın. Bu ürünlerde belge bitiş tarihi, EBS başvurusu, ÜTS talebi, Kurum teyidi ve geçiş süresi birlikte izlenmeli.
- Toplu işlem yapmadan önce veri doğrulama yapın. Excel hazırlamak sürecin son adımı olmalı, ilk adımı değil.
En büyük risk: ÜTS kaydını sadece portal işlemi sanmak
Bu kılavuzun verdiği en büyük mesaj şudur: ÜTS kaydı portalda birkaç alan doldurmak değildir. ÜTS kaydı, ürün verisinin, belge verisinin, firma rolünün, mevzuat rejiminin ve başvuru durumunun birlikte yönetildiği bir uyum operasyonudur.
Firmalar bunu sadece "kim ÜTS’ye girecek?" sorusuyla yönetirse eksik kalır. Asıl soru şu olmalı: Ürün başvuruya gerçekten hazır mı? Bağlı belge doğru mu? Belge ürün kapsamını gerçekten kapsıyor mu? Ürün sınıfı doğru mu? Temel UDI-DI var mı? EMDN doğru mu? Geçiş hükmü varsa kanıtı nerede? ÜTS başvuru sonucu takip ediliyor mu?
Bu sorulara sistematik cevap veremeyen firmalarda süreç kişilere bağımlı hale gelir. Kişi değiştiğinde bilgi kaybolur, Excel güncelliğini yitirir, belge tarihi kaçırılır ve başvuru gecikir.
Sonuç
Yeni ÜTS kayıt kılavuzu, tıbbi cihaz firmalarına açık bir mesaj veriyor: Ürün kaydı, belge yönetimi ve mevzuat uyumu artık ayrı ayrı yönetilecek işler değil. Hepsi aynı operasyonel zincirin parçası.
Bu zincirde bir halka eksikse, ürün kaydı gecikir. Belge yanlışsa, başvuru sorun yaşar. Sınıf yanlışsa, iş kuralı yanlış çalışır. Geçiş hükmü takip edilmezse, ürünün piyasadaki durumu belirsizleşir. Toplu Excel kontrol edilmezse, hata tek üründen yüzlerce ürüne yayılır.
Bu nedenle firmaların yeni dönemde yapması gereken şey çok net: ürün verisini temizlemek, belge envanterini ürünlerle eşleştirmek, MDR ve IVDR geçişlerini ürün bazında takip etmek ve ÜTS kayıt sürecini manuel takipten çıkarıp sistematik hale getirmek.
Bu kılavuz, doğru okunduğunda sadece bir mevzuat metni değil; tıbbi cihaz firmaları için ürün verisi yönetimi, belge disiplini ve operasyonel olgunluk çağrısıdır.