
EUDAMED
EUDAMED Türkiye’de Medikal Firmalar İçin Ne Anlama Geliyor? TİTCK Duyurusu Işığında ÜTS, MDR, IVDR ve Aktör Kaydı Yol Haritası
28 Mayıs 2026’da zorunlu hale gelecek ilk EUDAMED modülleri, ÜTS süreçlerine etkisi, aktör kaydı, UDI ve Cihaz kaydı, ithalatçı ve dağıtıcı ayrımı için Türkiye odaklı rehber.
- Yazar
- UTSBridge Ekibi
- Yayın tarihi
- 18 Mayıs 2026
- Okuma süresi
- 12 dk okuma

Bu yazı bir tanıtım metni değildir. Türkiye’deki tıbbi cihaz firmalarının EUDAMED gündemine girerken karşılaşacağı pratik soruları ve yol haritasını derli toplu bir biçimde toplar. Mevzuatın resmi metninin yerine geçmez; ekiplerin ortak dil kurmasına ve uyum çalışmalarını planlamasına yardımcı olur.
Avrupa Birliği tıbbi cihaz pazarında işler artık yalnızca CE belgesi almakla bitmiyor. MDR, IVDR, UDI, EUDAMED, Authorised Representative, ithalatçı, vigilance, piyasaya arz sonrası gözetim gibi kavramlar günlük operasyonun parçası haline geliyor. Türkiye’deki firmalar açısından konu daha da kritik: çünkü Türkiye pazarı ÜTS ile ayrı bir ulusal takip sistemine sahip. Avrupa tarafında ise EUDAMED devreye giriyor. Bu iki yapı birbirinin aynısı değil, ancak veri mantığı, ürün kayıt disiplini, üretici ve ithalatçı sorumlulukları açısından sürekli birbirine temas ediyor.
EUDAMED zorunluluk takvimi
TİTCK 2026/KKB-1 duyurusunun temel tarih çerçevesi aşağıdaki şemada özetlenmiştir. Bu takvim hem yeni piyasaya arz edilecek cihazlar için hem de halihazırda piyasada bulunan eski (legacy) cihazlar için kritik adımları içerir.
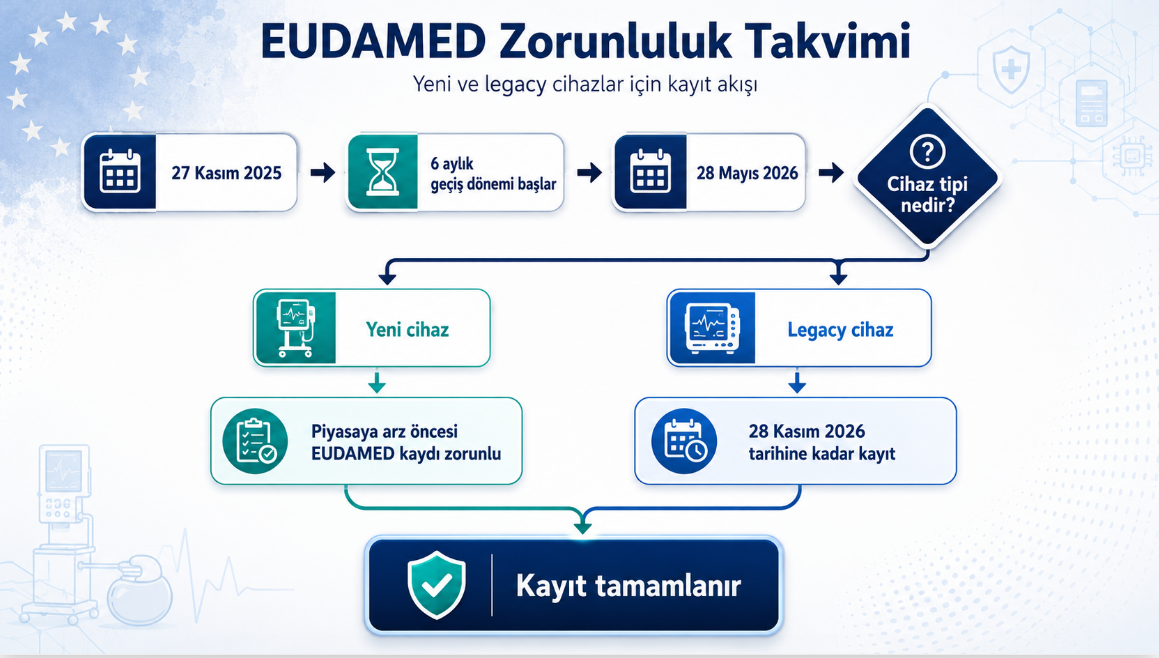
- 27 Kasım 2025: İlk dört modülün işlevselliği ilan edildi.
- 28 Mayıs 2026: Aktör Kaydı, UDI ve Cihaz Kaydı, Onaylanmış Kuruluşlar ve Sertifikalar, Piyasa Gözetimi modülleri zorunlu hale geliyor.
- 28 Mayıs 2026 sonrası: İlk satış birimi bu tarihte veya sonrasında piyasaya arz edilecek cihazlar, piyasaya arzdan önce EUDAMED’e kaydedilmelidir.
- 28 Kasım 2026: 28 Mayıs 2026’dan önce piyasaya arz edilmiş ve aynı UDI-DI ile devam eden legacy cihazlar için kayıt son tarihi.
- Old devices: UDI ve Cihaz Kaydı modülüne kaydedilmez.
EUDAMED kuralın değil sistemin adıdır
EUDAMED, Avrupa Birliği’nin tıbbi cihazlar için kurduğu merkezi veri tabanı ve dijital kayıt sistemidir. Tek bir portal değildir. Firma kaydı, cihaz kaydı, UDI bilgileri, sertifikalar, vigilance kayıtları, klinik araştırmalar ve piyasa gözetimi gibi süreçleri aynı ekosistemde toplar.
Çerçeveyi tek cümleyle netleştirmek gerekirse: MDR ve IVDR kural kitabıdır, EUDAMED bu kuralların işlendiği dijital sistemdir, ÜTS ise Türkiye’nin ulusal kayıt ve takip sistemidir. MDR ve IVDR, "ne yapmalısın?" sorusuna cevap verir. EUDAMED, "bu bilgileri nerede ve nasıl yöneteceksin?" sorusunu çözer. ÜTS ise Türkiye pazarı için paralel bir disiplin yürütür.
Asıl önemi firmayı veri disiplinine zorlamasıdır
EUDAMED’in önemi yalnızca Avrupa pazarı için bir kayıt zorunluluğu olmasından gelmiyor. Asıl önemi, tıbbi cihaz firmalarını çok daha disiplinli veri yönetimine zorlamasıdır. Birçok firmada ürün bilgisi farklı sistemlerde farklı şekilde tutulur. ERP’de ürün adı başka, ÜTS’de başka, CE belgesinde başka, teknik dosyada başka, etikette başka olabilir. Eski dönemde bu dağınıklık bir şekilde yönetilebiliyordu; ancak MDR, IVDR ve EUDAMED dünyasında ciddi risk üretir.
EUDAMED ekosistemi firmadan dolaylı olarak şunları ister:
- Ürün kimliği net olmalı: aynı cihaz farklı sistemlerde farklı isimle dolaşmamalı.
- UDI yapısı tutarlı olmalı: Basic UDI-DI, UDI-DI ve varyant ilişkisi bozulmamalı.
- Üretici, ithalatçı, yetkili temsilci ve dağıtıcı rolleri doğru tanımlanmalı.
- Sertifikalar, cihaz kayıtları ve teknik dosya birbiriyle çelişmemeli.
- Piyasaya arz sonrası izleme ve bildirim süreçleri kayıt altına alınabilmeli.
Bu yüzden EUDAMED hazırlığı sadece regulatory affairs ekibinin işi değildir. Ürün yönetimi, kalite, operasyon, IT, satın alma, satış, ithalat, ihracat ve üst yönetim birlikte düşünmek zorundadır.
Aktör kavramı firmanın hangi rol ile girdiğidir
EUDAMED’de aktör, tıbbi cihazın piyasaya arz zincirinde regülasyonel sorumluluğu olan taraftır. Aktör kavramını basitçe "firma" gibi düşünmemek gerek. Esas mesele firmanın hangi rolle sisteme girdiğidir.
- Manufacturer, üretici
- Authorised Representative, yetkili temsilci
- Importer, ithalatçı (AB veya Türkiye uyum mevzuatına göre tanımlı)
- System veya procedure pack producer, SİP imalatçısı
Aşağıdaki şema her firmanın kendine sorması gereken temel kararı özetler: aktör kaydı gerekli mi, gerekiyorsa hangi rol veya rollerle?
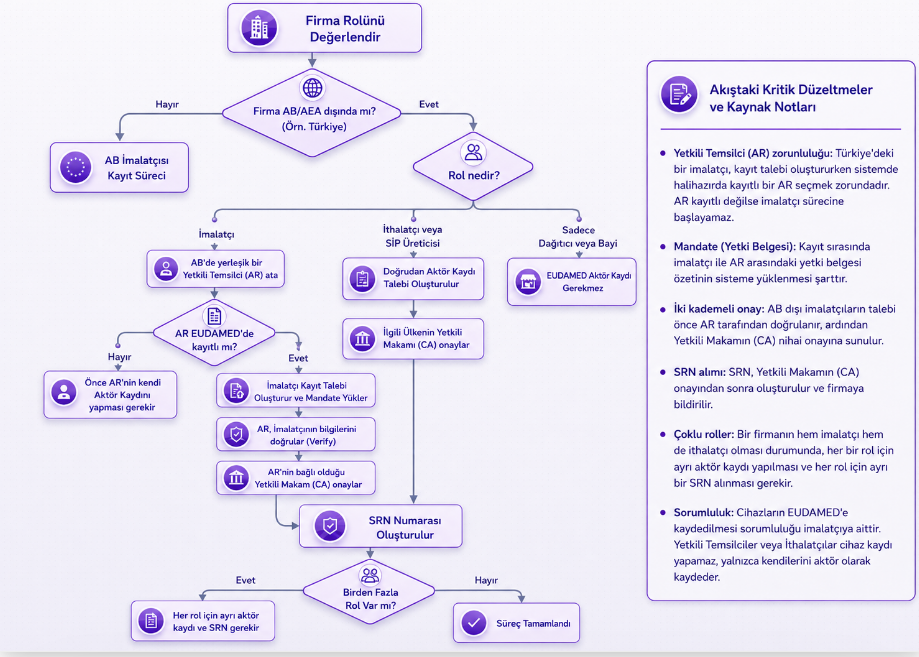
Türk firması EUDAMED’de nasıl aktör olur?
Türkiye’de yerleşik imalatçı, yetkili temsilci, ithalatçı veya SİP imalatçısı EUDAMED’de aktör kaydı başvurusu yaptığında, başvuru Türkiye’deki yetkili otorite olan TİTCK tarafından değerlendirilir ve onaylanır. Başvuru uygun bulunursa EUDAMED tarafından SRN veya Actor ID oluşturulur. Başvuruda eksiklik varsa düzeltme istenir, uygun değilse gerekçesiyle reddedilir.
AB ve Türkiye dışında yerleşik bir imalatçının başvurusu ise ilgili yetkili temsilci tarafından doğrulandıktan sonra yetkili otorite değerlendirmesine gider. Bu nedenle Türkiye’deki firmalar için "EUDAMED aktör kaydını mutlaka AB’deki yetkili temsilcinin bulunduğu ülkenin otoritesi onaylar" gibi genel bir ifade doğru değildir. Türkiye’de yerleşik firmaların aktör kaydını TİTCK onaylar.
TİTCK’nin rolü: Aktör kaydında onay mercii, cihaz verisinde doğrudan doğrulayıcı değil
TİTCK, Türkiye’deki aktör kayıt başvurularını değerlendirir ve onaylar. Ancak UDI ve Cihaz Kaydı modülüne girilen cihaz verilerinin doğrulanmasında yetkili otoritelerin doğrudan rolü bulunmaz. ÜTS’de ise cihaz ve SİP verilerinin doğruluğu ayrıca Kurum tarafından kontrol edilir. Bu nedenle EUDAMED ve ÜTS verileri çelişirse, öncelikle ÜTS verileri dikkate alınır.
Sistemde kim hangi veriyi girer?
EUDAMED’de herkes her veriyi girmez, roller ayrıdır. Üretici kendi cihaz verisinden, ürün kimliğinden, UDI bilgilerinden ve kayıt içeriğinin doğruluğundan sorumludur. Yetkili temsilci, özellikle EU 27, İzlanda, Lihtenştayn, Norveç, Türkiye veya Kuzey İrlanda dışında yerleşik üreticiler için devreye giren temsil ilişkisidir. Bu nedenle Türkiye’de yerleşik bir üreticinin EUDAMED’de üretici aktör kaydı yapması, aktör kartında mutlaka ayrıca yetkili temsilci görünmesini gerektiren bir varsayım olarak ele alınmamalıdır. İthalatçı cihaz kaydı oluşturmaz; kendi ithalatçı rolü kapsamında ilgili üreticiyle aktör seviyesinde bağlantı kurar ve üreticinin girdiği verileri kontrol eder. Onaylanmış Kuruluş, sertifika ve uygunluk değerlendirme süreçlerine ilişkin kayıtları işler veya doğrular. Yetkili otorite ise aktör kayıtlarını onaylar, piyasa gözetimi ve denetim süreçlerinde devreye girer; EUDAMED cihaz verisini ÜTS’deki gibi tek tek baştan doğrulayan makam olarak düşünülmemelidir.
Aşağıdaki şema cihaz kaydında kim ne yapar sorusunu görselleştirir.
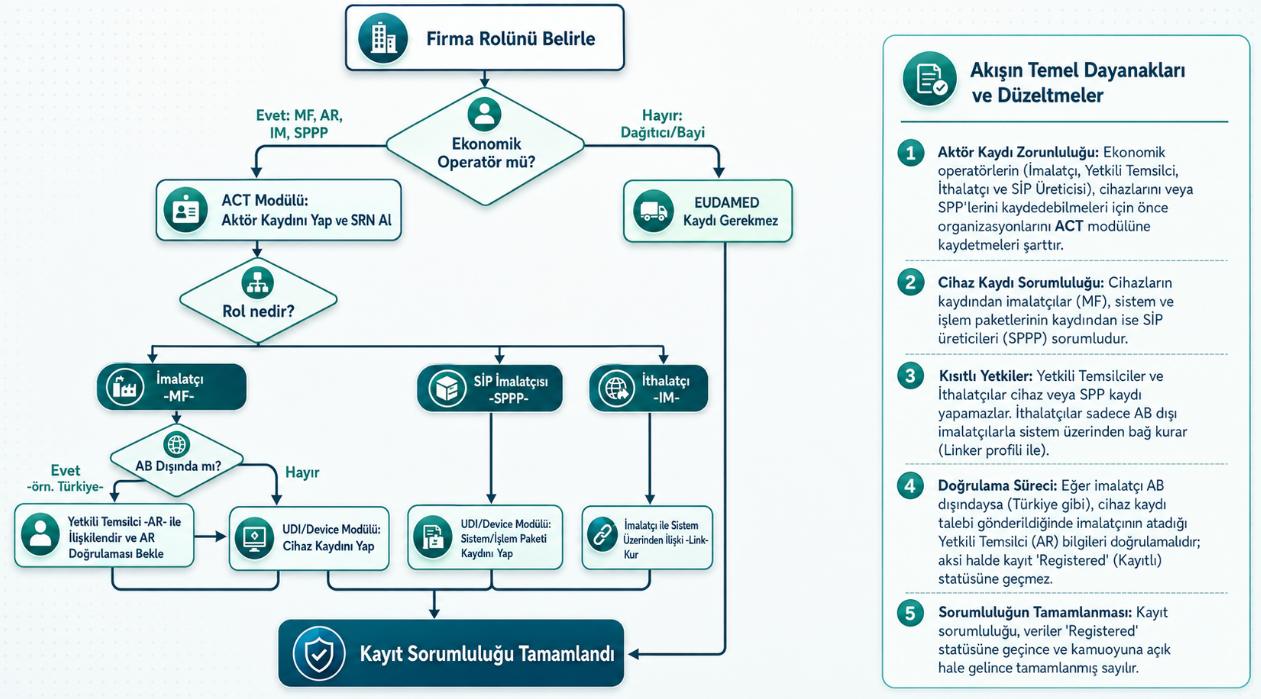
ÜTS’de ithalatçı olmak, EUDAMED’de importer olmak değildir
AB mevzuatına göre ithalatçı, bir cihazı üçüncü bir ülkeden AB piyasasına arz eden AB’de yerleşik gerçek veya tüzel kişidir. Türkiye uyum mevzuatında bu tanım Türkiye pazarı için uyarlanmıştır. Bu nedenle sadece AB’de yerleşik bir imalatçının cihazlarını Türkiye’ye ithal eden firmalar, AB mevzuatı açısından dağıtıcı kabul edilir ve EUDAMED’de ithalatçı olarak aktör kaydı yapmalarına gerek yoktur.
EUDAMED UDI ve Cihaz Kaydı neyi içerir, neyi içermez?
EUDAMED’de cihazlar ve SİP’ler cihaz tanımlayıcısına göre kaydedilir. Bu tanımlayıcı UDI-DI veya EUDAMED DI olabilir. Lot numarası, seri numarası, üretim tarihi gibi UDI-PI bilgileri UDI ve Cihaz Kaydı modülüne kaydedilmez.
Bu ayrım önemlidir. EUDAMED cihaz kimliği ve UDI-DI düzeyinde kayıt disiplini getirir. Lot ve seri izlenebilirliği ise ÜTS, stok, hareket ve firma içi operasyonlarda kritik olmaya devam eder.
Belge ve sertifika doğruluğu kimde?
EUDAMED cihaz kaydı, ÜTS’deki gibi doğrudan otorite tarafından cihaz verisinin tek tek doğrulandığı bir süreç olarak düşünülmemelidir. Aktör kaydı yetkili otorite tarafından onaylanır; cihaz verisinin doğruluğu ise öncelikle ilgili iktisadi işletmecinin sorumluluğundadır. Sertifika tarafında Notified Body kayıtları ve uygunluk değerlendirme süreçleri ayrıca önem taşır.
Kontrol birkaç katmanda işler. Aktör kayıt aşamasında yetkili otorite firmanın gerçekten var olduğunu ve rolünün doğru tanımlandığını inceler. Notified Body katmanında MDR veya IVDR kapsamındaki cihazların sertifikaları üzerinde gerçek karşılığı olmayan kayıtlar tutarsızlık üretir. Yetkili temsilci katmanı, AB veya Türkiye dışında yerleşik üreticinin teknik dosya ve uygunluk beyanı bilgilerini kontrol eder. Piyasa gözetimi ve denetim ayağında ise yetkili otorite, gümrük, hastane, rakip şikayeti, vigilance bildirimi veya denetim sonucunda belge doğruluğu yeniden sorgulanabilir.
MDR ile EUDAMED arasındaki fark
MDR, Avrupa Birliği’nin genel tıbbi cihaz mevzuatıdır. Sınıflandırma, piyasaya arz, klinik değerlendirme, UDI, teknik dosya, piyasaya arz sonrası gözetim ve vigilance gibi kuralları belirler. EUDAMED ise MDR ve IVDR kapsamındaki bilgilerin kaydedildiği ve yönetildiği dijital altyapıdır.
Kısaca, MDR kuraldır, EUDAMED sistemdir. MDR, "bu ürünü nasıl belgelendireceksin?" der. EUDAMED, "bu ürüne ait kayıt, UDI, aktör, sertifika ve bildirimleri nerede yöneteceksin?" der.
IVDR hangi ürünleri bağlar?
IVDR, in vitro tanı cihazlarını bağlar. Yani insan vücudundan alınan örneklerin vücut dışında analiz edilmesiyle tıbbi bilgi üreten ürünler IVDR kapsamına girebilir. Tipik örnekler:
- Kan test kitleri
- PCR testleri
- HIV, hepatit, Covid testleri
- Gebelik testleri
- Genetik test kitleri
- Laboratuvar reaktifleri
- Kalibratör ve kontrol materyalleri
- Tanısal amaçlı yazılımlar
Kritik soru: ürün insan örneğinden tanı, risk, sağlık durumu veya tedavi kararı için bilgi üretiyor mu? Cevap evet ise IVDR ihtimali güçlenir. Yine de laboratuvarda kullanılan her ürün IVDR değildir. Sadece araştırma amaçlı (RUO işaretli) veya tanı sonucu üretmeyen ürünler IVDR dışında kalabilir. Her ürün kendi intended purpose, label claim ve kullanım amacına göre değerlendirilmelidir.
MDR ve IVDR ile eski belgelendirmeden ne değişti?
MDR ve IVDR, MDD ve IVDD dönemine göre daha sıkı, daha veri odaklı ve daha izlenebilir bir yapı getirdi. MDR tarafında klinik değerlendirme, UDI, post-market surveillance, PMCF, ekonomik operatör sorumlulukları ve teknik dosya yapısı güçlendi.
IVDR’de değişim daha serttir. IVDD döneminde birçok in vitro tanı ürünü üreticinin kendi beyanıyla piyasaya sunulabiliyordu. IVDR ile risk sınıflandırması A, B, C, D olarak yeniden kuruldu. Çok daha fazla ürün Notified Body denetimine girdi.
ÜTS ile EUDAMED arasındaki ilişki
ÜTS, Türkiye’nin ulusal tıbbi cihaz kayıt ve takip sistemidir. EUDAMED, AB’nin merkezi tıbbi cihaz veri tabanıdır. Biri diğerinin yerine geçmez. Türkiye’de piyasaya arz için ÜTS yükümlülükleri devam eder. AB pazarına giriş veya AB mevzuatıyla bağlantılı süreçlerde EUDAMED ve MDR ile IVDR tarafı önem kazanır.
Bu çerçevede sorun çok net: firmalar bugün iki ayrı sistemi, aynı ürün verisiyle ve tutarlı şekilde yönetmek zorunda. Aynı ürün için ÜTS’de bir veri seti, ERP’de başka bir ürün kartı, CE dosyasında başka bilgiler, etikette başka ifadeler bulunabilir. EUDAMED bunların regülasyonel olarak tutarlı olmasını bekler.
Tek ürün verisi, çok sistemli kullanım
Doğru strateji şudur: ürün ana verisi tek merkezde yönetilmeli ve ÜTS, EUDAMED, ERP, kalite sistemi ile regülasyon dosyaları aynı veri disiplininden beslenmelidir.
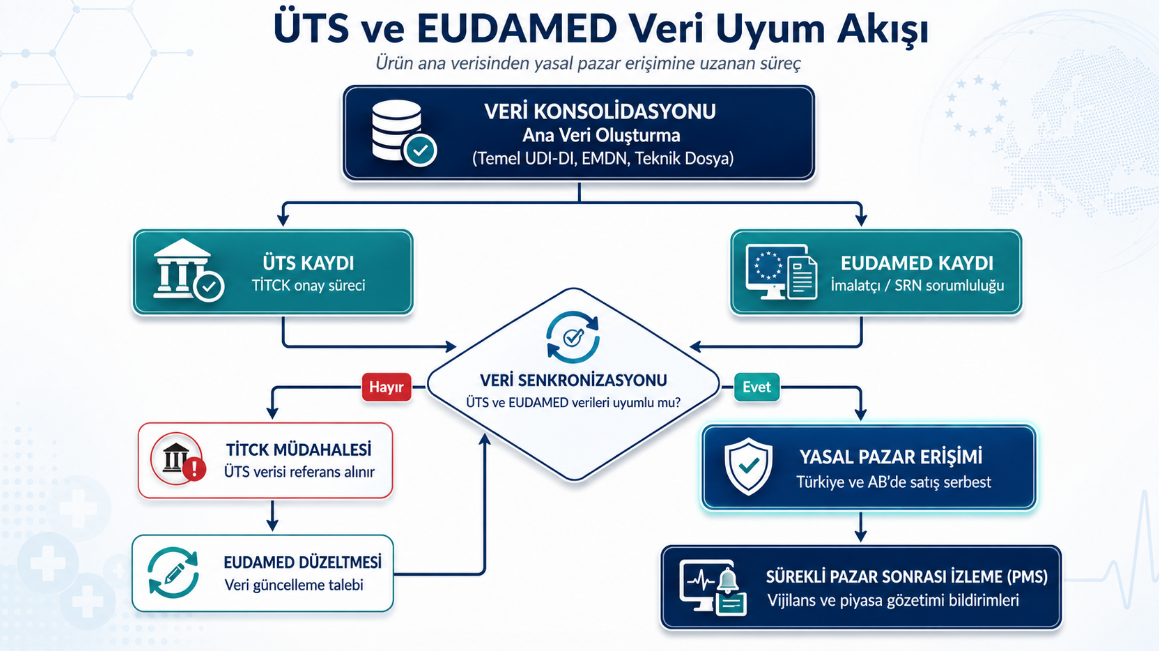
- Tek doğru kaynak (single source of truth) olarak ürün ana verisi merkezi kurulur.
- Bu merkezden ÜTS kayıtları beslenir, Türkiye pazarı operasyonu yürür.
- Aynı merkezden EUDAMED kayıtları beslenir, AB pazarı uyum süreci yürür.
- Aynı merkez ERP veya stok sistemi ile senkron tutulur, operasyonel veri uyumu sağlanır.
- Sertifika ve doküman yönetimi de bu merkeze bağlanır; belge ile cihaz ilişkisi bozulmaz.
Türkiye’de firmalar nerede zorlanacak?
EUDAMED hazırlığında Türkiye’deki firmaların en çok zorlanacağı alanın mevzuat okumak değil, veri düzeni kurmak olduğunu söylemek mümkündür. Özellikle riskli alanlar şunlardır:
- Ürün ana verisinin dağınık olması; aynı ürünün farklı sistemlerde farklı tanımlanması.
- GTIN, UDI, Basic UDI-DI ve ürün varyant hiyerarşisinin net olmaması.
- Üretici, ithalatçı, distribütör, yetkili temsilci ve EUDAMED importer rollerinin karıştırılması.
- CE sertifikası, DoC, etiket ve ÜTS bilgilerinin birbiriyle uyuşmaması.
- MDR ve IVDR ayrımının ürün bazında net yapılmaması.
- Sertifika geçerlilik tarihleri ve Notified Body ilişkilerinin manuel takip edilmesi.
- Çok ürünlü firmalarda toplu veri hazırlığının Excel üzerinden kontrolsüz yürütülmesi.
- Vigilance ve piyasaya arz sonrası izleme süreçlerinin ürün master data ile bağlı olmaması.
Bu sorunlar küçük görünür ancak EUDAMED yükleme, denetim veya ihracat sürecinde büyük operasyon krizine dönüşebilir.
Firmalar bugünden ne yapmalı? Readiness yol haritası
EUDAMED hazırlığı için firmaların bugünden bir readiness programı başlatması gerekir. Aşağıdaki sıralı adımlar pratik bir başlangıç çerçevesi sunar.
- Rol analizi: firma ürün bazında manufacturer mı, importer mı, distributor mü, AB veya Türkiye dışında yerleşik üretici mi, yetkili temsilci ilişkisi var mı netleştirilir.
- Aktör rol doğrulaması: ÜTS’deki firma rolü ile EUDAMED ve MDR rolü karşılaştırılır. Firma gerçekten imalatçı mı, ithalatçı mı, dağıtıcı mı, SİP imalatçısı mı netleştirilir.
- Ürün envanteri: hangi ürün MDR, hangisi IVDR, hangisi legacy device, hangisi aktif veya pasif, hangisinin sertifikası riskte gibi başlıklar çıkarılır.
- MDR ve IVDR ayrımı: her ürün için intended purpose, sınıf ve regulation kapsamı doğrulanır.
- UDI ve master data kontrolü: Basic UDI-DI, UDI-DI, GTIN, EMDN, risk sınıfı tek tek doğrulanır.
- Sertifika ve doküman eşleştirme: CE sertifikası, DoC, teknik dosya, etiket, kullanım kılavuzu, ÜTS kaydı ve ERP kartı aynı ürünle birebir eşleşmelidir.
- ÜTS ve EUDAMED uyuşmazlık kontrolü: ÜTS’deki ürün adı, barkod, belge, üretici, ithalatçı, marka ve cihaz sınıfı bilgileri EUDAMED’e hazırlanacak veriyle karşılaştırılır. Çelişki varsa önce ÜTS verisi referans alınarak düzeltme planı yapılır.
- Readiness skoru: her ürünün yayına hazır mı, blocking eksiği var mı raporu çıkar.
- Veri girişi planı: portal, XML toplu yükleme veya M2M entegrasyon kararı verilir.
Yönetim ekipleri konuyu nasıl görmeli?
EUDAMED hazırlığı yalnızca bir regulatory compliance maliyeti gibi görülmemelidir. Doğru yönetilirse şirketin operasyon kalitesini yükselten bir dönüşüm programına dönüşür. Bu çalışma sonucunda firma şu kazanımları elde eder:
- Daha temiz ürün ana verisi
- Daha kontrollü ÜTS operasyonu
- Sertifika ve belge risklerinin erken görülmesi
- AB pazarına girişte daha az sürpriz
- Denetimlere daha hazırlıklı yapı
- Satış, kalite ve regülasyon ekipleri arasında daha az çelişki
- Recall, FSCA veya vigilance gibi kritik durumlarda daha hızlı hareket kabiliyeti
EUDAMED yalnızca bir yük değildir. Doğru kurulduğunda güven, hız ve rekabet avantajıdır.
UTSBridge bu süreçte nerede konumlanır?
EUDAMED ve ÜTS birlikte düşünüldüğünde, firmaların asıl ihtiyacı yalnızca bir başvuru ekranı değildir. Asıl ihtiyaç, ürün ana verisini, belge ilişkilerini, UDI bilgilerini, MDR ve IVDR ayrımını, üretici, ithalatçı ve dağıtıcı rollerini ve ÜTS kayıt durumunu aynı disiplin içinde yönetebilmektir.
UTSBridge bu noktada Türkiye’deki medikal firmalar için operasyonel bir hazırlık katmanı olarak konumlanır. Ürün ana verisinin düzenlenmesi, ÜTS kayıt süreçlerinin takibi, belge ve sertifika ilişkilerinin görünür hale getirilmesi, MDR ve IVDR bazlı ürün ayrımı, eksik veri kontrolü ve ÜTS ile EUDAMED’e hazırlanacak veri setleri arasındaki tutarlılık analizi gibi alanlarda firmalara daha kontrollü bir çalışma zemini sağlar.
Bu yaklaşım, EUDAMED’e doğrudan veri gönderimi vaadinden önce, daha temel bir problemi çözer: firmanın elindeki ürün ve belge verisinin doğru, tutarlı, denetlenebilir ve yeniden kullanılabilir hale gelmesi. Çünkü ÜTS ve EUDAMED tarafında sürdürülebilir uyumun başlangıç noktası, temiz ve güvenilir ürün ana verisidir.